Преждевременное старение
Прогерия Хатчинсона-Гилфорда - Hutchinson-Gilford progeria syndrome (HGPS)
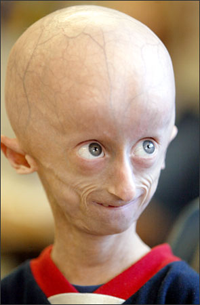 Редкое генетическое заболевание, характеризуемое преждевременным старением и увеличением числа сердечных заболеваний. Синдром описан впервые в 1886 году. С тех пор зафиксировано более 100 случаев его проявления. Синдром проявляется в среднем у одного из 8 миллионов новорожденных.
Редкое генетическое заболевание, характеризуемое преждевременным старением и увеличением числа сердечных заболеваний. Синдром описан впервые в 1886 году. С тех пор зафиксировано более 100 случаев его проявления. Синдром проявляется в среднем у одного из 8 миллионов новорожденных.
Первые проявления заболевания появляются в 12-14 месяцев и заключаются в потере волос и отставании в росте. Кроме того, прогерия характеризуется появлением морщинистой кожи, преждевременного атеросклероза, остеопороза, деформаций костей, что выражается в диспропорциях черепа и лица, недоразвитии нижней челюсти и ключицы, смещении бедер, недоразвитие зубов и других патологий (рис.1).
У больных детей начинают проявляться болезни старческого периода, однако их разум развивается в соответствии с их реальным возрастом,
так что их старение нельзя считать абсолютно аналогичным старению здорового человека. Дети с HGPS умирают в среднем в 12-13 лет, обычно от сердечных приступов или кровоизлияний в мозг и, как правило, живут от 8 до 21 года.
Ген LMNA кодирует предшественник ламина А
HGPS возникает из-за мутации одного гена LMNA, кодирующего преламин А/C, приводящей к делеции 50 аминокислот в предшественнике белка ламинаА -преламине А, который является структурным белком ядерной мембраны (рис.2, 3).
В нормальных клетках ген LMNA состоит из 25000 пар нуклеотидов, мРНК преламина А состоит из 12 экзонов. В седьмом экзоне кодируется сигнал ядерной локализации (NLS), необходимый для транспортировки белка из цитоплазмы в ядро. В десятом экзоне находится сайт альтернативного сплайсинга ламин А/ламин С. Белок преламина А состоит из 664 аминокислот и несет на С-конце CAAX-мотив (661-664), в котором С-цистеин, А - остаток обычно алифатической
аминокислоты, Х может быть любым остатком. Такой мотив также обнаружен в ламине B1, B2 и у многих других клеточных белков. CAAX мотив запускает три последовательных ферментативных реакции посттрансляционной модификации, приводящие к зрелому ламину А, структурному белку ядерной мембраны:
1. Фарнезилтрансфераза (FTase) добавляет липид С15-фарнезил к тиольной группе цистеина в пределах CCAX мотива.
2.Следующие три аминокислоты (т.е. AAX) удаляются белокпрениловой специфической эндопротеазой. Расщепление белка производят две мембранные эндопротеазы эндоплазматического ретикулума, Zmpste24 and Rce1
3. Вновь образованный фарнезилцистеин метилируется пренилбелковой специфической метилтрансферазой эндоплазматического ретикулума Icmt.
После того как модификации CAAX мотива выполнены, преламин А (в отличие от других CAAX-содержащих белков) подвергается дополнительной модификации. Фрагмент С-концевых 15 аминокислот
(647-661) белка, вместе с метилированным фарнезилцистеином, вырезается металлопротеазой Zmpste24 и деградирует. Оставшая часть является зрелым ламином А (рис.4). Фарнезилирование преламина А важно для узнавания ядерной мембраны. Каждая из трех модификаций преламина А создает гидрофобность С-конца, обеспечивая ассоциацию его с внутренней ядерной мембраной, после чего белок расщепляется.

рис 2. Схема строения гена LMNA

рис 3. Схема строения мРНК и белка преламина А. UTR - нетранслируемый регион, NLS - сигнал ядерной локализации, CAAX - мотив пренилирования.
G608G, E145K и R471C, R527C - мутации приводящие к HGPS (показаны зеленым).
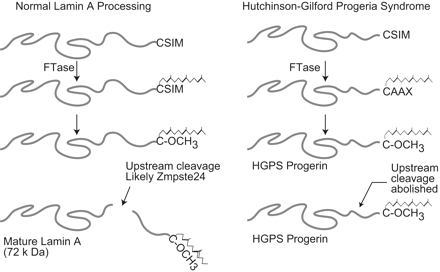
рис 4.
Мутантный ламин
Наиболее часто HGPS происходит при точечных мутациях экзона 11 LMNA, мутация происходит в кодоне 608 и активирует загадочный сайт сплайсинга, приводящий к делеции в 50 аминокислот в рамке считывания преламина А (606-656 аминокислоты), но при этом остается С-концевой CAAX мотив. При этом теряется сайт по которому происходит расщепление белка преламина во время созревания. В нормальных клетках преламин обнаружить не удается, так как он сразу же переводится в зрелый ламин А. Мутантный преламин А, образующийся при HGPS, который называют также прогерин, вмещает CAAX мотив, запускающий фарнезилирование, но делеция в 50 аминокислот предотвращает последующий процессинг в зрелый ламин А, что приводит к накоплению прогерина в нуклеоплазме ядра. Это приводит к нестабильности мембраны: нарушается ее
целостность, появляются характерные выпячивания, увеличивается пористость, что приводит к ускорению гибели клеток.
В экспериментах на культурах клеток фибробластов было показано, что при недостатке Zmpste24 количество преламина А увеличивалось и появлялись изменения характерные для клеток больных прогерией - ядра фибробластов деформированы и содержат многочисленные пузыри.
Преламин А токсичен и уменьшение его уровня по крайней мере до 50% обеспечивает защиту от заболевания HGPS, что может помочь в поиске подхода к лечению этого синдрома.
- S.H. Yang, M.O. Bergo , J.I. Toth, X. Qiao, Y. Hu, S. Sandoval, M. Meta , P. Bendale , M.H. Gelb , S.G. Young, L.G. Fong Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. PNAS, 2005.
- A.B. Csoka, H. Cao, P. J. Sammak, D. Constantinescu,
G. P. Schatten and R. A. Hegele Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes Journal of Medical Genetics 2004; 41:304-308 - B.C. Capell , M.R. Erdos, J.P.Madigan, J. J. Fiordalisi, R. Varga, K. N. Conneely, L.B. Gordon, C.J. Der, A.D. Cox and F.S. Collins, Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome PNAS, 2005, no. 36, 12879-12884.
- L.G. Fong, J.K. Ng , M.Meta, N. Cote, S. H. Yang, C.L. Stewart, T. Sullivan, A. Burghardt, S. Majumdar, K. Reue, Martin O. Bergo and S.G. Young Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice PNAS, 2004, no. 52, 18111-18116.